The paper, Pain
Most recent blogposts:
Most recent blogposts:
Part 14: Side trip out to the periphery! Part 14b: Prevention of pain neurotags is WAY easier than cure Part 14c: PW Nathan was an interesting pain researcher Part 14d: Brain glia are from neuroectoderm and PNS glia are from neural crest Part 14e: The stars in our headsPart 14f: Gleeful about glia Part 14g: ERKs and MAPKs and pain Part 14h: glia-fication of nociceptive input 14i: molecular mediators large and small Part 14j: Neurons, calling glia (over, do you read?) Part 14k: Glia calling glia, over. Do you read?
SEE ALL PREVIOUS BLOGPOSTS IN THIS SERIES LISTED AT THE END
Yup, hard to believe, but we're still on our fogbound island waiting for clarity, cracking the brain open with that gliopathy paper, awaiting the opportunity to go back to the Melzack and Katz paper. I thought it was going to be a fast, easy trip floating down a delightful river, but as it turned out, we went upstream, and furthermore, there have been not only meanders, but some unexpected dives, twists, turns and now this long delay.
I'm pretty sure in the end it will have done the brain a lot of good to have tackled this information. If nothing else, it might be easier, next time around.
I'm pretty sure in the end it will have done the brain a lot of good to have tackled this information. If nothing else, it might be easier, next time around.
Anyway, today we plow through what goes on in the peripheral ganglia, DRG and trigeminal, how satellite cells converse with neurons.

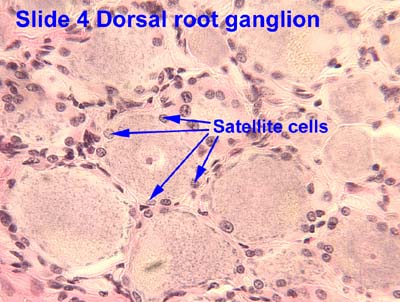 |
| Satellite cells in DRG - the little dark cells clustered between neurons SOURCE |
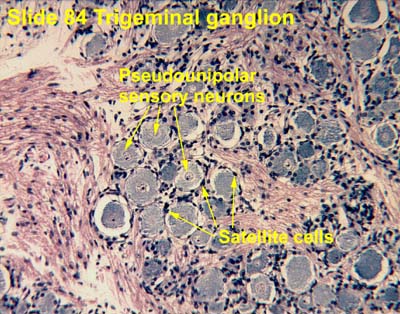 |
| Same story in trigeminal ganglion SOURCE |
Satellite cells connect with each other in the DRG via gap junctions. Remember those? Connexins etc? Gap junction communication becomes "greatly enhanced in persistent pain conditions." I.e., gap junctions appear, and gap junction communication increases, between SGCs and neurons in the DRG.
- Nerve is injured, or at least acts like it has been
- local inflammation
- Puringergic signalling happens. i.e., activity-dependent ATP escapes from the (supposedly injured) neuronal cell's cell body
- P2X7 is activated in satellite cell
- This stimulates production of TNF-alpha by satellite cell..
- ..which increases the excitability of surrounding neurons.
Remember, with gap junctions, actual stuff inside the cell can cross over into another cell.
Sounds like they start to lose their individuality, their integrity - cell boundaries are breached. Or broached. Or both.
But wait, there's more:
Satellite cells make ATP too. It activates P2X3 receptor expressed in neurons. One of those many feed-forward cycles in physiology that amplifies nociception, this one via peripheral sensitization.
But wait - there's even more, and it has to do with morphine analgesia.
Morphine elicits MMP-9, one of those "ase"-s (ases tear stuff down). MMP-9 is elicited by production of μ-opioid receptors. μ-opioid receptors form when morphine is supplied. If MMP-9 is knocked out, analgesia by morphine is no longer "masked." I think that means the cell can't make the tear-down molecule so the morphine analgesia works better.
Morphine stimulates production of GFAP (to make scar) and IL-1 beta. Because MMP-9 stimulates production of both, apparently. Anyway, IL-1 beta activates receptors for itself in neurons. Which helps them make action potentials. As if they needed help for that. It enhances sodium currents and suppresses potassium currents. Masks morphine analgesia in its own fascinating and creepy little ways.
Anyway, the authors say, targeting interactions between satellite cells and neurons in the DRGs and TGs can enhance opioid analgesia.
Morphine elicits MMP-9, one of those "ase"-s (ases tear stuff down). MMP-9 is elicited by production of μ-opioid receptors. μ-opioid receptors form when morphine is supplied. If MMP-9 is knocked out, analgesia by morphine is no longer "masked." I think that means the cell can't make the tear-down molecule so the morphine analgesia works better.
Morphine stimulates production of GFAP (to make scar) and IL-1 beta. Because MMP-9 stimulates production of both, apparently. Anyway, IL-1 beta activates receptors for itself in neurons. Which helps them make action potentials. As if they needed help for that. It enhances sodium currents and suppresses potassium currents. Masks morphine analgesia in its own fascinating and creepy little ways.
Anyway, the authors say, targeting interactions between satellite cells and neurons in the DRGs and TGs can enhance opioid analgesia.
..............
There is yet another section, glial mediators modulating excitatory and inhibitory synaptic transmission. Included are these points:
- Very low concentrations will do it
- For frank excitation we have proinflammatory cytokines and chemokines, TNF-alpha, IL-1beta, IFN-gamma, CCL2, TSP4, and enhanced signalling of glutamate receptors postsynaptically.
- TNF-apha, IL-1beta, IL-6 elicit longterm neuronal plasticity in the "pain" circuit (yeah, they really do use that word)
- For reduction or loss of normal inhibition, we have reduced expression of KCC2, messing with anion gradients, which can turn a normally inhibitory anionic synaptic current into an excitatory one.
- BDNF, cyytokines, chemokines, PGE2 can mess with synaptic transmission
- Normal GABA and glycine currents can become suppressed by IL1beta, IL-6
- ATP, morphine-stimulated microglia can help BDNF be pumped out
- Enhancement of inhibition is not well understood. Yet. IL-10 can maybe suppress TNF-alpha somehow, details still not worked out.
- NPD1 (neuroprotectin) and RvE1 (resolvin) can block TNF-alpha somehow. [I see a whole new line of heroic action figures in the future..]
Finally, after all these days, here are the authors' conclusions re: gliopathy, what it is.
- Astrocytes lose stop maintaining homeostasis of potassium and glutamate, and neurons become more excitable
- Astrocytic water channel AQP4 becomes dysfunctional. Edema results in the CNS and PNS.
- Glia can't insulate the "pain" circuit anymore. [Yes, they still use the p word.]
- Instead, glia amplify "pain", by producing pronociceptive and proinflammatory mediators
- So, the storyline is, neuron is injured, signals its signal. Glia make their stuff. The stuff activates more stuff. The second batch of stuff, unleashed, makes more pathways and facilitates more cell-cell communication. But they can also make antinflammatory and antinociceptive stuff, so..
Anyway, they leave off with suggestions for further trials. Most of the current information comes from rodent glia studies. Because human glia are so much bigger and more complex than rodent glia, human glia should be way more carefully studied using current up-to-the-minute methods and technology.
They don't really answer their own question, they just ask it.
My hunch: It could be that all this is all just business as usual, not glia being sick in and of themselves, necessarily.
Which leaves a door wide, wide open for plenty of possibility, for descending modulation from rostral centres to come down, via hundreds-fold more neurons, regulate all these synapses in the dorsal horn, tell the neurons to shush, tell the glia to stop being so danged hyperexciting, regain control of the anion gradients, change the concentration gradients of interleukins and BDNF, etc., boost the production of anti-nociceptive and anti inflammatory molecules, get it turned around, get the central poles of those pseudounipolar neurons that comprise the DGRs to tell their cell bodies to stop bothering the satellite cells, wait for the satellite cells to stop being bothered and go back to normal, and get it all to decrease, like water rolling down a hill.
I think we're ready to sail away from here. Fog is lifted. Back to Melzack and Katz!
A very nice pier from which to re-embark.
 |
| SOURCE |
.................
Previous blogposts
Previous blogposts
Part 1 First two sentences Part 2 Pain is personal Also Pain is Personal addendum., Neurotags! Pain is Personal, Always.
Part 3a Pain is more than sensation: Backdrop Part 3b Pain is not receptor stimulation Part 3c: Pain depends on everything ever experienced by an individual
Part 4: Pain is a multidimensional experience across time
Part 5: Pain and purpose
Part 6a: Descartes and his era; Part 6b: History of pain - what’s in “Ref 4”?; Part 6c: History of pain, Ref 4, cont.. : There is no pain matrix, only a neuromatrix; Part 6d: History of Pain: Final takedown Part 6e: Pattern theories in the history of pain Part 6f: Evaluation of pain theories Part 6g: History of Pain, the cautionary tale. Part 6h: Gate Control Theory.
Part 7: Gate control theory has stood the test of time: Patrick David Wall; Part 7b: Gate control: "The theory was a leap of faith but it was right!"
Part 8: Beyond the gate: Self as mayor Part 8b: 3-ring circus of self Part 8c: Getting objective about subjectivity
Part 9: Phantom pain - in the brain! Part 9b: Dawn of the Neuromatrix model Part 9c: Neuromatrix: MORE than just spinal projection areas in thalamus and cortex Part 9d: More about phantom body pain in paraplegics
Part 10: "We don't need a body to feel a body." Part 10b: Conclusion1: The brain generates its own experience of being in a body Part 10c:Conclusion 2: Your brain, not your body, tells you what you're feeling Part 10d: Conclusion 3: The brain's sense of "Self" can INclude missing parts, or EXclude actual parts, of the biological body Part 10e: The neural network that both comprises and moves "Self" is (only)modified by sensory experience
Part 11: We need a new conceptual brain model! Part 11b: Intro to a new conceptual nervous system Part 11c: Older brain models just don't cut it Part 11d: The NEW brain model!
Part 7: Gate control theory has stood the test of time: Patrick David Wall; Part 7b: Gate control: "The theory was a leap of faith but it was right!"
Part 8: Beyond the gate: Self as mayor Part 8b: 3-ring circus of self Part 8c: Getting objective about subjectivity
Part 9: Phantom pain - in the brain! Part 9b: Dawn of the Neuromatrix model Part 9c: Neuromatrix: MORE than just spinal projection areas in thalamus and cortex Part 9d: More about phantom body pain in paraplegics
Part 10: "We don't need a body to feel a body." Part 10b: Conclusion1: The brain generates its own experience of being in a body Part 10c:Conclusion 2: Your brain, not your body, tells you what you're feeling Part 10d: Conclusion 3: The brain's sense of "Self" can INclude missing parts, or EXclude actual parts, of the biological body Part 10e: The neural network that both comprises and moves "Self" is (only)modified by sensory experience
Part 11: We need a new conceptual brain model! Part 11b: Intro to a new conceptual nervous system Part 11c: Older brain models just don't cut it Part 11d: The NEW brain model!
Part 11: We need a new conceptual brain model! Part 11b: Intro to a new conceptual nervous system Part 11c: Older brain models just don't cut it Part 11d: The NEW brain model!
No comments:
Post a Comment